Publications
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Full list (since 2012) |
|
|
|
|
|
|
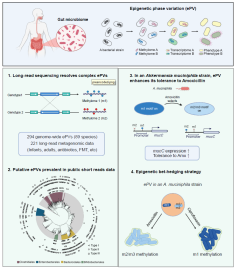
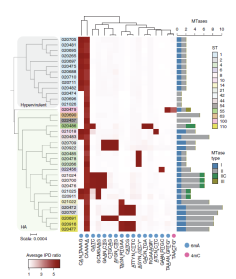
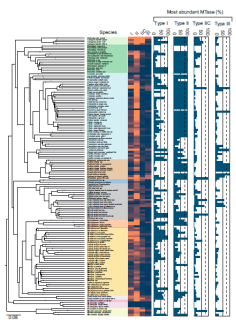
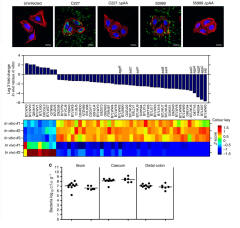
Epigenetic phase variation in the gut microbiome
enhances bacterial adaptation |
|
|
|
|
|
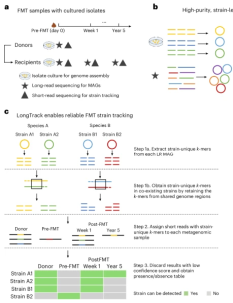
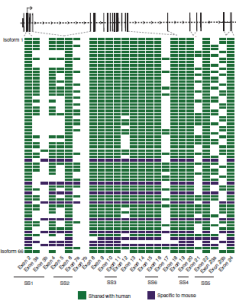
Long-read metagenomics for strain tracking after faecal
microbiota transplant |
|
|
|
|
|
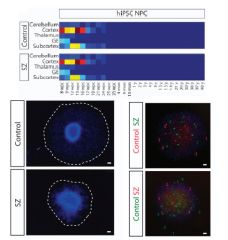
Phenotypic complexities of rare heterozygous neurexin-1
deletions |
|
|
|
|
|
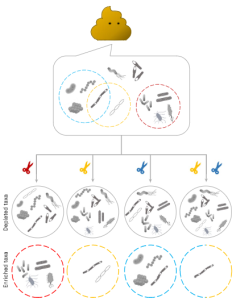
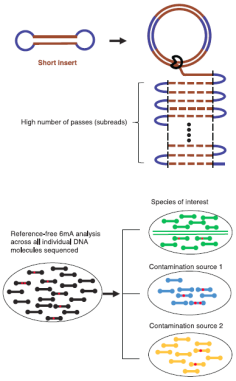
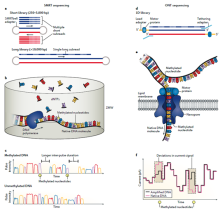
mEnrich-seq: methylation-guided enrichment sequencing of
bacterial taxa of interest from microbiome |
|
|
|
|
|
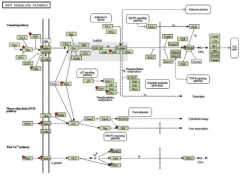
Navigating the pitfalls of mapping DNA and
RNA modifications |
|
|
|
|
|
Critical assessment of DNA adenine
methylation across eukaryotes using
quantitative deconvolution |
|
|
|
|
|
Discovering
multipletypes of DNA methylation from
individual bacteria and microbiome using
nanopore sequencing |
|
|
|
|
|
Epigenomic
characterization of Clostridioides
difficile finds a conserved DNA
methyltransferase that mediates
sporulation and pathogenesis Media coverage: GenomeWeb, Technology Networks, Medical News, PHYS, MEDPAGE Today, PacBio Blog. |
|
|
|
|
|
Conserved
DNA Methyltransferases: A Window into
Fundamental Mechanisms of Epigenetic
Regulation in Bacteria |
|
|
|
|
|
Neuronal
impact of patient-specific aberrant NRXN1α
splicing Media coverage: ScienceDaily, Medical News, Medical Express, PacBio Blog. |
|
|
|
|
|
Deciphering
bacterial epigenomes using modern
sequencing technologies |
|
|
|
|
|
Metagenomic
binning and association of plasmids with
bacterial host genomes using DNA
methylation Highlighted in Nature Methods (link) Media coverage: GEN News, PacBio, GenomeWeb, MD Magazine, BioITWorld, Science Daily, Infection Control Today, PHYS. |
|
|
|
|
|
Mapping
and characterizing N6-methyladenine in
eukaryotic genomes using single molecule
real-time sequencing N6-methyladenine
(m6dA) has been discovered as a novel form of
DNA methylation prevalent in eukaryotes,
however, methods for high resolution mapping
of m6dA events are still lacking.
Single-molecule real-time (SMRT) sequencing
has enabled the detection of m6dA events at
single-nucleotide resolution in prokaryotic
genomes, but its application to detecting m6dA
in eukaryotic genomes has not been rigorously
examined. Herein, we identified unique
characteristics of eukaryotic m6dA methylomes
that fundamentally differ from those of
prokaryotes. Based on these differences, we
describe the first approach for mapping m6dA
events using SMRT sequencing specifically
designed for the study of eukaryotic genomes,
and provide appropriate strategies for
designing experiments and carrying out
sequencing in future studies. We apply the
novel approach to study two eukaryotic
genomes. For green algae, we construct the
first complete genome-wide map of m6dA at
single nucleotide and single molecule
resolution. For human lymphoblastoid cells
(hLCLs), it was necessary to integrate SMRT
sequencing data with independent sequencing
data. The joint analyses suggest putative m6dA
events are enriched in the promoters of young
full-length LINE-1 elements (L1s), but call
for validation by additional methods. These
analyses demonstrate a general method for
rigorous mapping and characterization of m6dA
events in eukaryotic genomes. |
|
|
|
|
|
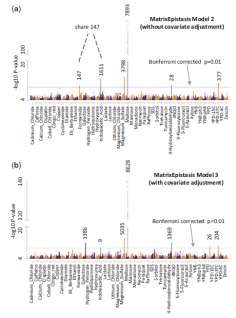
MatrixEpistasis:
ultrafast, exhaustive epistasis scan for
quantitative traits with covariate
adjustment |
|
|
|
|
|
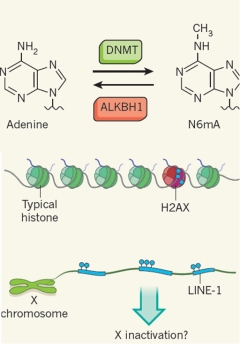
DNA
methylation on N6-adenine in mammalian
embryonic stem cells Tao P. Wu, Tao Wang, Matthew G. Seetin, Yongquan Lai, Shijia Zhu, Kaixuan Lin, Yifei Liu, Stephanie D. Byrum, Samuel G. Mackintosh, Mei Zhong, Alan Tackett, Guilin Wang, Lawrence S. Hon, Gang Fang, James Swenberg & Andrew Xiao It has been
widely accepted that 5-methylcytosine is the
only form of DNA methylation in mammalian
genomes. Here we identify N6-methyladenine as
another form of DNA modification in mouse
embryonic stem cells. Alkbh1 encodes a
demethylase for N6-methyladenine. An increase
of N6-methyladenine levels in Alkbh1-deficient
cells leads to transcriptional silencing.
N6-methyladenine deposition is inversely
correlated with the evolutionary age of LINE-1
transposons; its deposition is strongly
enriched at young (<1.5 million years old)
but not old (>6 million years old) L1
elements. The deposition of N6-methyladenine
correlates with epigenetic silencing of such
LINE-1 transposons, together with their
neighbouring enhancers and genes, thereby
resisting the gene activation signals during
embryonic stem cell differentiation. As young
full-length LINE-1 transposons are strongly
enriched on the X chromosome, genes located on
the X chromosome are also silenced. Thus,
N6-methyladenine developed a new role in
epigenetic silencing in mammalian evolution
distinct from its role in gene activation in
other organisms. Our results demonstrate that
N6-methyladenine constitutes a crucial
component of the epigenetic regulation
repertoire in mammalian genomes. |
|
|
|
|
|
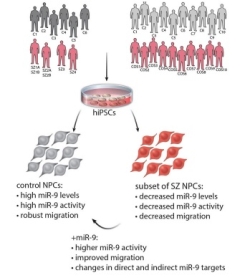
Dysregulation
of miRNA-9 in a Subset of Schizophrenia
Patient-Derived Neural Progenitor Cells Aaron Topol*, Shijia Zhu*, Brigham J. Hartley, Jane English, Mads E. Hauberg, Ngoc Tran, Chelsea Ann Rittenhouse, Anthony Simone, Douglas M. Ruderfer, Jessica Johnson, Ben Readhead, Yoav Hadas, Peter A. Gochman, Ying-Chih Wang, Hardik Shah, Gerard Cagney, Judith Rapoport, Fred H. Gage, Joel T. Dudley, Pamela Sklar, Manuel Mattheisen, David Cotter, Gang Fang# & Kristen J. Brennand# Converging
evidence indicates that microRNAs (miRNAs) may
contribute to disease risk for schizophrenia
(SZ). We show that microRNA-9 (miR-9) is
abundantly expressed in control neural
progenitor cells (NPCs) but also significantly
downregulated in a subset of SZ NPCs. We
observed a strong correlation between miR-9
expression and miR-9 regulatory activity in
NPCs as well as between miR-9 levels/activity,
neural migration, and diagnosis.
Overexpression of miR-9 was sufficient to
ameliorate a previously reported neural
migration deficit in SZ NPCs, whereas
knockdown partially phenocopied aberrant
migration in control NPCs. Unexpectedly,
proteomic- and RNA sequencing (RNA-seq)-based
analysis revealed that these effects were
mediated primarily by small changes in
expression of indirect miR-9 targets rather
than large changes in direct miR-9 targets;
these indirect targets are enriched for
migration-associated genes. Together, these
data indicate that aberrant levels and
activity of miR-9 may be one of the many
factors that contribute to SZ risk, at least
in a subset of patients. |
|
|
|
|
|
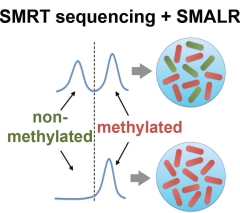
Single
molecule-level detection and long read-based
phasing of epigenetic variations in bacterial
methylomes John Beaulaurier, Xue-Song Zhang, Shijia Zhu, Robert Sebra, Chaggai Rosenbluh, Gintaras Deikus, Nan Shen, Diana Munera, Matthew K Waldor, Martin J Blaser, Andrew Chess, Eric E Schadt#, Gang Fang# Beyond its role
in host defense, bacterial DNA methylation
also plays important roles in the regulation
of gene expression, virulence and antibiotic
resistance. Bacterial cells in a clonal
population can generate epigenetic
heterogeneity to increase population-level
phenotypic plasticity. Single molecule,
real-time (SMRT) sequencing enables the
detection of N6-methyladenine and
N4-methylcytosine, two major types of DNA
modifications comprising the bacterial
methylome. However, existing SMRT
sequencing-based methods for studying
bacterial methylomes rely on a
population-level consensus that lacks the
single-cell resolution required to observe
epigenetic heterogeneity. Here, we present
SMALR (single-molecule modification analysis
of long reads), a novel framework for single
molecule-level detection and phasing of DNA
methylation. Using seven bacterial strains, we
show that SMALR yields significantly improved
resolution and reveals distinct types of
epigenetic heterogeneity. SMALR is a powerful
new tool that enables de novo detection of
epigenetic heterogeneity and empowers
investigation of its functions in bacterial
populations. |
|
|
|
|
|
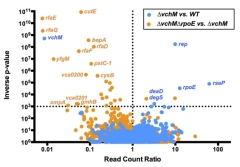
A
Cytosine Methytransferase Modulates the Cell
Envelope Stress Response in the Cholera
Pathogen Michael C. Chao, Shijia Zhu, Satoshi Kimura, Brigid M. Davis, Eric E. Schadt, Gang Fang,# Matthew K. Waldor# Methylation of
DNA is used by numerous organisms to regulate
a wide variety of cellular processes, but
specific roles for most DNA methyltransferases
have not been defined. We studied one such
enzyme in Vibrio cholerae, the cholera
pathogen, using genome-wide approaches to
compare DNA methylation, gene expression, and
the sets of genes required or dispensable for
growth in bacterial strains that produced or
lacked this enzyme. These studies allowed us
to identify numerous cellular processes
regulated, either directly or indirectly, by
this cytosine methyltransferase. In
particular, we found that an absence of enzyme
activity was associated with reduced levels of
a bacterial stress response; consequently, a
stress response pathway that is essential in
wild type bacteria is not needed for survival
of the mutant lacking the methyltransferase.
Similar genome-wide analyses can likely to be
used to define the cellular roles of many
additional uncharacterized DNA
methyltransferases. |
|
|
|
|
|
Autotransporters
but not pAA are critical for rabbit
colonization by Shiga toxin-producing
Escherichia coli O104:H4 Diana Munera, Jennifer M. Ritchie, Stavroula K. Hatzios, Rod Bronson, Gang Fang, Eric E. Schadt, Brigid M. Davis & Matthew K. Waldor The outbreak of
diarrhoea and haemolytic uraemic syndrome that
occurred in Germany in 2011 was caused by a
Shiga toxin-producing enteroaggregative
Escherichia coli (EAEC) strain. The strain was
classified as EAEC owing to the presence of a
plasmid (pAA) that mediates a characteristic
pattern of aggregative adherence on cultured
cells, the defining feature of EAEC that has
classically been associated with virulence.
Here we describe an infant rabbit-based model
of intestinal colonization and diarrhoea
caused by the outbreak strain, which we use to
decipher the factors that mediate the
pathogen's virulence. Shiga toxin is the key
factor required for diarrhoea. Unexpectedly,
we observe that pAA is dispensable for
intestinal colonization and development of
intestinal pathology. Instead,
chromosome-encoded autotransporters are
critical for robust colonization and
diarrhoeal disease in this model. Our findings
suggest that conventional wisdom linking
aggregative adherence to EAEC intestinal
colonization is false for at least a subset of
strains. |
|
|
|
|
|
Altered WNT
Signaling in Human Induced Pluripotent Stem
Cell Neural Progenitor Cells Derived from Four
Schizophrenia Patients Aaron Topol, Shijia Zhu, Ngoc Tran, Anthony Simone, Gang Fang, Kristen J. Brennand Schizophrenia
(SZ) is a devastating psychiatric disorder
hypothesized to be a neurodevelopmental
condition arising as a consequence of
dysregulation of brain development. WNT
signaling is important for neural patterning,
proliferation and migration, and synapse
formation; converging postmortem, rodent, and
pharmacologic evidence suggests that WNT
signaling may contribute to SZ. We used human
induced pluripotent stem cell (hiPSC) derived
forebrain patterned neural progenitor cells
(NPCs) to investigate canonical WNT activity
in a pilot cohort of four patients with SZ.
Future studies comprising larger patient
cohorts are necessary to determine whether
aberrant canonical WNT signaling is a causal
molecular factor contributing to aberrant
neural patterning and neuronal maturation in
SZ or simply a noncell autonomous consequence
of increased oxidative stress. |
|
|
|
|
|
Phenotypic
differences in hiPSC NPCs derived from
patients with schizophrenia Kristen Brennand, Jeffrey Savas, Yongsung Kim, Ngoc Tran, Anthony Simone, Kazue Hashimoto-Torii, Kristin Beaumont, Hyung Joon Kim, Aaron Topol, Ian Ladran, Mohammed Abdelrahim, Bridget Matikainen-Ankney, Shih-hui Chao, Milan Mrksich, Pasko Rakic, Gang Fang, Bin Zhang, John Yates III, Fred H. Gage Consistent with
recent reports indicating that neurons
differentiated in vitro from human-induced
pluripotent stem cells (hiPSCs) are immature
relative to those in the human brain, gene
expression comparisons of our hiPSC-derived
neurons to the Allen BrainSpan Atlas indicate
that they most resemble fetal brain tissue.
This finding suggests that, rather than
modeling the late features of schizophrenia
(SZ), hiPSC-based models may be better suited
for the study of disease predisposition. We
now report that a significant fraction of the
gene signature of SZ hiPSC-derived neurons is
conserved in SZ hiPSC neural progenitor cells
(NPCs). We used two independent
discovery-based approaches—microarray gene
expression and stable isotope labeling by
amino acids in cell culture (SILAC)
quantitative proteomic mass spectrometry
analyses—to identify cellular phenotypes in SZ
hiPSC NPCs from four SZ patients. From our
findings that SZ hiPSC NPCs show abnormal gene
expression and protein levels related to
cytoskeletal remodeling and oxidative stress,
we predicted, and subsequently observed,
aberrant migration and increased oxidative
stress in SZ hiPSC NPCs. These reproducible
NPC phenotypes were identified through
scalable assays that can be applied to
expanded cohorts of SZ patients, making them a
potentially valuable tool with which to study
the developmental mechanisms contributing to
SZ. |
|
|
|
|
|
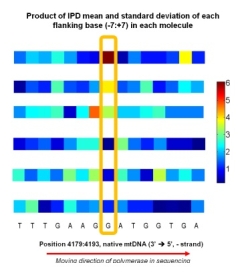
Modeling Kinetic Rate Variation in Third
Generation DNA Sequencing Data to Detect
Putative Modifications to DNA Bases Eric E. Schadt*, Onureena Banerjee*, Gang Fang*, Zhixing Feng, Wing H. Wong, Xuegong Zhang, Andrey Kislyuk, Tyson A. Clark, Khai Luong, Vipin Kumar, Alice Chen-Plotkin, Neal Sondheimer, Jonas Korlach, Andrew Kasarskis. While
significant inroads have been made identifying
small nucleotide variation and structural
variations in DNA that impact phenotypes of
interest, progress has not been as dramatic
regarding epigenetic changes and base-level
damage to DNA, largely due to technological
limitations in assaying all known and unknown
types of modifications at genome scale.
Recently single molecule real time (SMRT)
sequencing has been reported to identify
kinetic variation (KV) events that have been
demonstrated to reflect epigenetic changes of
every known type, providing a path forward for
detecting base modifications as a routine part
of sequencing. However, to date, no
statistical framework has been proposed to
enhance the power to detect these events while
also controlling for false positive events. By
modeling enzyme kinetics in the neighborhood
of an arbitrary location in a genomic region
of interest as a conditional random field, we
provide a statistical framework for
incorporating kinetic information at a test
positions of interest as well as at
neighboring sites that help enhance the power
to detect KV events. The performance of this
and related models is explored, with the best
performing model applied to plasmid DNA
isolated from Escherichia coli and
mitochondrial DNA isolated from human brain
tissue. We highlight widespread kinetic
variation events, some of which strongly
associate with known modification events while
others represent putative chemically modified
sites of unknown types. |
|
|
|
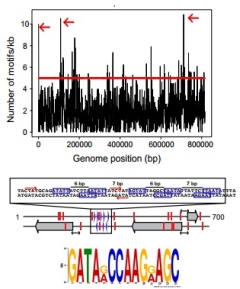 |
Comprehensive
methylome characterization of Mycoplasma
genitalium and Mycoplasma pneumoniae, at
single-base resolution Maria Lluch Senar, Khai Luong, Veroica Llorens, Javi Delgado, Gang Fang, Kristi Spittle, Tyson Clark, Eric Schadt, Steve Turner, Jonas Korlach, Luis Serrano We define the
methylome of two closely related bacteria, M.
genitalium and M. pneumoniae, by
single-molecule real-time (SMRT) DNA
sequencing. In M. pneumoniae we found two
previously unknown N6-methyladenine
methyltransferase specificities, one of which
is also found in M. genitalium. The common
methyltransferase is a Dam-like methylase, and
was attributed to its corresponding gene using
cloned plasmids in a methyltransferase-free E.
coli strain, while the second methylase is of
type I and uniquely present in M. pneumoniae.
Analysis of the distribution of methylation
sites across the genome of M. pneumoniae at
exponential and stationary growth suggests a
potential role for methylation in regulating
the cell cycle as well as in gene regulation.
|
|
|
|
 |
Detecting
DNA modifications from SMRT sequencing data by
modeling sequence context dependence of
polymerase kinetic Zhixing Feng, Gang Fang, Jonas Korlach, Tyson Clark, Khai Luong, Xuegong Zhang, Wing Wong, and Eric Schadt DNA modications
such as methylation and DNA damage can play
critical regulatory roles in biological
systems. Single molecule, real time (SMRT)
sequencing technology generates DNA sequences
as well as DNA polymerase kinetic information
that can be used for the direct detection of
DNA modications. We demonstrate that local
sequence context has a strong impact on DNA
polymerase kinetics in the neighborhood of the
incorporation site during the DNA synthesis
reaction, allowing for the possibility of
estimating the expected kinetic rate of the
enzyme at the incorporation site using kinetic
rate information collected from existing SMRT
sequencing data (historical data) covering the
same local sequence contexts of interest. We
develop a Empirical Bayesian hierarchical
model for incorporating historical data. Our
results show that the model could greatly
increase DNA modication detection accuracy,
and reduce requirement of control data
coverage. For some DNA modications that have a
strong signal, a control sample is even not
needed by using historical data as alternative
to control. Thus, sequencing cost can be
greatly reduced by using the model. |
|
|
|
|
|
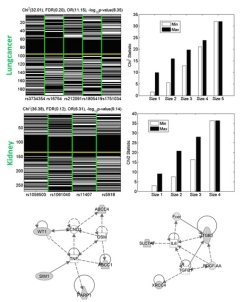
High-order
SNP Combinations Associated with Complex
Diseases: Efficient Discovery, Statistical
Power and Functional Interactions Gang Fang*, Majda Haznadar, Wen Wang, Haoyu Yu, Michael Steinbach, Tim Church, William Oetting, Brian Van Ness and Vipin Kumar*. There has been
increased interest in discovering combinations
of single-nucleotide polymorphisms (SNPs) that
are strongly associated with a phenotype even
if each SNP has little individual effect.
Efficient approaches have been proposed for
searching two-locus combinations from
genome-wide datasets. However, for high-order
combinations, existing methods either adopt a
brute-force search which only handles a small
number of SNPs (up to few hundreds), or use
heuristic search that may miss informative
combinations. In addition, existing approaches
lack statistical power because of the use of
statistics with high degrees-of-freedom and
the huge number of hypotheses tested during
combinatorial search. We designed an efficient
and effective framework for high-order
combinations in case-control datasets. The
substantially improved efficiency and
scalability demonstrated on synthetic and real
datasets with several thousands of SNPs allows
the study of several important mathematical
and statistical properties of SNP combinations
with order as high as eleven. We further
explore functional interactions in high-order
combinations and reveal a general connection
between the increase in discriminative power
of a combination over its subsets and the
functional coherence among the genes
comprising the combination, supported by
multiple datasets. Finally, we study several
significant high-order combinations discovered
from a lung-cancer dataset and a
kidney-transplant-rejection dataset in detail
to provide novel insights on the complex
diseases. Interestingly, many of these
associations involve combinations of common
variations that occur in small fractions of
population. Thus, our approach is an
alternative methodology for exploring the
genetics of rare diseases for which the
current focus is on individually rare
variations. |
|
|
|
 |
Mining
Low-support Discriminative Patterns from Dense
and High-dimensional Data Gang Fang, Gaurav Pandey, Wen Wang, Manish Gupta, Michael Steinbach and Vipin Kumar. Discriminative
patterns can provide valuable insights into
data sets with class labels, that may not be
available from the individual features or the
predictive models built using them. Most
existing approaches work efficiently for
sparse or low-dimensional data sets. However,
for dense and high-dimensional data sets, they
have to use high thresholds to produce the
complete results within limited time, and
thus, may miss interesting low-support
patterns. In this paper, we address the
necessity of trading off the completeness of
discriminative pattern discovery with the
efficient discovery of low-support
discriminative patterns from such data sets.
We propose a family of antimonotonic measures
named SupMaxK that organize the set of
discriminative patterns into nested layers of
subsets, which are progressively more complete
in their coverage, but require increasingly
more computation. In particular, the member of
SupMaxK with K ¼ 2, named SupMaxPair, is
suitable for dense and high-dimensional data
sets. Experiments on both synthetic data sets
and a cancer gene expression data set
demonstrate that there are low-support
patterns that can be discovered using
SupMaxPair but not by existing approaches.
Furthermore, we show that the low-support
discriminative patterns that are only
discovered using SupMaxPair from the cancer
gene expression data set are statistically
significant and biologically relevant. This
illustrates the complementarity of SupMaxPair
to existing approaches for discriminative
pattern discovery. |
|
|
|
|
|
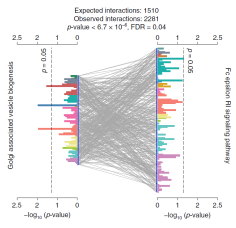
Discovering
genetic interactions bridging pathways in
genome-wide association studies |
|
|
|
|
|
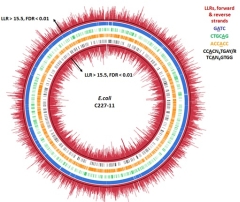
Genome-wide
map of methylated adenine residues using
single-molecule real-time sequencing in
pathogenic Escherichia coli Gang Fang, Diana Munera, David I. Friedman, Anjali Mandlik, Michael C. Chao, Onureena Banerjee, Zhixing Feng, Bojan Losic, Milind C. Mahajan, Omar J. Jabado, Gintaras Deikus, Tyson A. Clark, Khai Luong, Iain A. Murray, Brigid M. Davis, Alona Keren-Paz, Andrew Chess, Richard J. Roberts, Jonas Korlach, Steve W. Turner, Vipin Kumar, Matthew K. Waldor, Eric E. Schadt Single-molecule
real-time (SMRT) DNA sequencing allows the
systematic detection of chemical modifications
such as methylation but has not previously
been applied on a genome-wide scale. We used
this approach to detect 49,311 putative
6-methyladenine (m6A) residues and 1,407
putative 5-methylcytosine (m5C) residues in
the genome of a pathogenic Escherichia coli
strain. We obtained strand-specific
information for methylation sites and a
quantitative assessment of the frequency of
methylation at each modified position. We
deduced the sequence motifs recognized by the
methyltransferase enzymes present in this
strain without prior knowledge of their
specificity. Furthermore, we found that
deletion of a phage-encoded
methyltransferase-endonuclease
(restriction-modification; RM) system induced
global transcriptional changes and led to gene
amplification, suggesting that the role of RM
systems extends beyond protecting host genomes
from foreign DNA. |
|
|
|